Medtronic Pipeline Flex Embolization Device
Medtronic is recalling nearly 9,000 Pipeline Flex embolization devices due to the potential for the device to break off during use. Broken pieces could enter the bloodstream and cause blockage, stroke, or death.
So far, the FDA has received reports of 59 malfunctions. There have been 10 serious injuries reported, and two deaths. This recall is a Class I, which is the most serious designation.
Philips CPAP and BiPAP Machines
Philips is recalling millions of CPAP and BiPAP machines due to foam inside the unit breaking down. Patients may inhale the particles or chemicals from the foam while using the device. The FDA has received patient reports of:
- Cough
- Chest Pressure
- Upper Airway Irritation
- Sinus Infection
Particulate exposure could cause more serious issues including asthma, inflammation, and even cancer due to carcinogenic chemicals.
Medtronic Septostomy Catheters
Medtronic and the FDA are recalling Medtronic Rashkind Balloon Septostomy Catheters due to a risk of breakage or device failure. Potential injuries include damage to the blood vessels or patient death. At least two deaths have been linked to the product.
BD Alaris System Infusion Pump
Becton Dickinson (BD) CareFusion 303 is recalling certain lots of the Alaris System Infusion Pump due to software and system errors. The recall affects 774,000 units.
The FDA reports 55 patients have been injured due to the errors, and one patient has died.

Medtronic HeartWare Ventricular Assist System (HVAD)
Medtronic is recalling certain HeartWare Ventricular Assist Systems (HVAD) due to the possibility of interruption in the electrical connection between the power source and controller.
The HeartWare heart pumps were originally recalled in 2018. In January 2020, Medtronic sent an Urgent Field Safety Notice to healthcare providers after receiving 36 complaints. At least one patient has died due to the device malfunctioning.

Applied Medical Embolectomy Catheters
The U.S. Food and Drug Administration (FDA) has announced a recall of Applied Medical Embolectomy catheters due to the tips detaching from the devices. The Class I recall includes:
- Python Embolectomy Catheters
- Bard Emobolectomy Catheters
- OTW Latis Cleaning Catheters
The catheters were originally recalled in October 2019. However, there have been at least 45 complaints since 2015. In May 2020, the FDA updated their recall information to include more than 19,000 devices.
Boston Scientific Angiographic Catheter
Boston Scientific has announced a recall of Imager II 5F Angiographic Catheters after receiving complaints about the tips detaching from the device. The U.S. Food and Drug Administration classified the recall as a Class I, meaning that using the device may cause serious injuries or death.
Detached tips can enter the blood vessel causing obstruction (embolism). Possible consequences include stroke or death.
More Information:
- https://www.fda.gov/medical-devices/medical-device-recalls/boston-scientific-corporation-recalls-imager-ii-angiographic-catheters-due-tip-detachment?utm_campaign=2020-04-06%20Boston%20Scientific%20Corporation%20Recalls%20Imager%20II%20Angiographic%20Catheters&utm_medium=email&utm_source=Eloqua
- https://www.druganddevicewatch.com/2020/04/17/boston-scientific-recall-class-fda/

Cardinal Health Surgical Gowns
The U.S. Food and Drug Administration (FDA) has announced a recall of Cardinal Health surgical gowns manufactured in China. According to the FDA, the gowns were made in facilities that do not maintain proper environmental conditions.
As a result, the gowns may not be sterile, and could pose a threat to patients in contact with them.

Mavidon Medical Devices
Mavidon has recalled all of their medical devices due to possible contamination with Burkholderia cepacia. The medical devices subject to this recall are skin-prepping products for medical and surgical use. The products include:
- LemonPrep
- PediaPrep
- Wave Prep
- Cardio Prep
- Collodions
- Collodion removers
Burkholderia cepacia is a pathogenic microorganism that can cause outbreaks of illness including respiratory infections.
More Information:
Zimmer Biomet ROSA Brain 3.0 Robotic Surgery System
The U.S. Food and Drug Administration (FDA) has announced a recall of Zimmer Biomet ROSA brain 3.0 robotic surgery systems. The recall is due to a possible problem with the software that could cause the robot to position itself incorrectly. Discrepancies in positioning can result in injury to the patient.
At the time of the recall, the FDA had received five complaints related to the software problem. At least one patient has been injured.
Medtronic 6 French Sherpa NX Active Guide Catheter
Medtronic is recalling more than 106,000 heart catheters due to the potential for the outer material to separate from the device. Separation could result in fragments entering the patient’s bloodstream.
Possible complications noted in the U.S. Food and Drug Administration (FDA) recall notice include blockage or injury to the blood vessel, blood clots, embolism, heart attack, and death.
At the time of the recall, the FDA had received at least five complaints.

Allergan - BIOCELL Textured Breast Implants
The U.S. Food and Drug Administration (FDA) requested that Allergan recall BIOCELL Textured Breast Implants and tissue expanders. The FDA requested all of these devices be removed from the global market after reports linked them to breast implant-associated anaplastic large cell lymphoma (BIA-ALCL).
There were more than 570 reports of BIA-ALCL, including 33 deaths. Of these reports, 481 women had the Allergan BIOCELL implants.
Integra LifeSciences - LimiTorr and MoniTorr Drainage Systems
Integra LifeSciences recalled LimiTorr and MoniTorr drainage systems after reports of pieces of the device breaking off. Device breakage could cause infection, backflow of air, subdural hematoma, over-drainage or death.
At the time of the recall, the U.S. Food and Drug Administration (FDA) had received serious adverse events reports.
ICU Medical, Inc. - Vial Spikes
ICU Medical, Inc. recalled certain lots of ChemoLock and ChemoClave Vial Spikes after burr particulate was found coming off of the protective cap. The burr particulate was determined to be a danger because it could detach and potentially enter the fluid path. This could subsequently lead to:
- Embolism
- Organ damage
- Stroke
- Death
The FDA classified the recall as a Class I – the most serious and threatening to patients.

Roche Diagnostics - CoaguChek Test Strips
Roche Diagnostics recalled certain lots of CoaguChek XS PT Test Strips due to inaccurate test results. The test strips were primarily used in patients taking blood thinning medications like Warfarin.
The inaccurate results posed the risk of life-threatening adverse events or death.

Zoll LifeVest 4000 Wearable Cardioverter Defibrillator
The Zoll LifeVest 4000 Wearable Cardioverter Defibrillator was recalled after the manufacturer discovered the device may be defective. The FDA reported that the device may fail to provide life-saving shock for patients with heart arrhythmia.
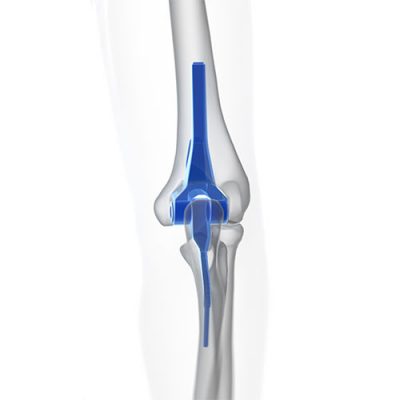
DePuy Synthes - Radial Head Prosthesis
Johnson & Johnson issued a recall of the DePuy Synthes radial head prosthesis due to
a risk of the component that anchors the prosthesis to the arum loosening. The device anchors to the radius bone in the arm, and if the connection becomes loose, the patient may experience:
- Pain
- Poor joint mechanics
- Post-operative fracture
- Soft tissue damage
- Osteolysis (pathological destruction of bone tissue)
Patients implanted with the device are encouraged to follow normal instructions and use. If symptoms present, patients should contact their healthcare provider immediately.
Smith & Nephew - Modular REDAPT™ Hip Systems
All lots of the Smith & Nephew Modular REDAPT™ Hip System were recalled after a higher than anticipated number of complaints. Patient complaints and adverse event trends included reports of corrosion at the neck stem, and the potential for metallosis (metal poisoning).
Patients reported the following symptoms:
- Pain
- Loosening of the device
- Device failure
- Need for revision surgeries
Patients experiencing symptoms should contact their healthcare provider immediately and schedule imaging and blood tests. Patients with questions can call 978-749-1440.

Stryker - LFIT Anatomic CoCr V40 Femoral Heads
A Class II recall was issued for Stryker LFIT v40 Femoral Head modular components after receiving numerous complaints about patient injuries. Patients reported adverse events including corrosion, spontaneous dislocation, and metallosis (metal poisoning). These defects resulted in complaints including:
- Swelling
- Pain
- Device failure
- Skin Rash
- Kidney and Liver Problems
- Tissue Death
- Additional Surgeries
Patients implanted with the recalled product are urged to contact their healthcare provider if they experience any of these symptoms. Following the recall, healthcare providers and facilities were asked to quarantine the devices pending return to the manufacturer.
MicroPort Orthopedics - Conserve, Dynasty, and Lineage Cups
Three MicroPort Orthopedics medical devices were recalled after rate trends indicated specific hazards and harm as a result of metal debris coming off of the devices. The metal debris reportedly causes adverse tissue reactions in the tissue surrounding the implant. The devices affected by the recall include:
- Conserve – Acetabular cup paired with Conserve cobalt chromium femoral head.
- Dynasty – Acetabular cup and metal liner paired with Conserve cobalt chromium femoral head.
- Lineage – Acetabular cup and metal liner paired with Lineage or Conserve cobalt
chromium femoral head.
The tissue reaction can cause patients to experience pain, loosening of the implant, device failure, and the need for additional surgeries. If metal debris enters the bloodstream, additional symptoms may occur, including rash, kidney and thyroid problems, and cardiomyopathy.
Contact your physician if you are experiencing signs and symptoms of complications. For questions regarding the recall, call 901-867-4771.

Ethicon Physiomesh Flexible Composite Hernia Mesh
Ethicon recalled Physiomesh Flexible Composite Hernia Mesh devices after research showed a much higher rate of hernia recurrence among patients. Following the recall, Ethicon removed the device from the market for laparoscopic hernia repair. The device remained on the market for traditional hernia repair surgery.

MicroPort Orthopedics - Profemur Stem
MicroPort Orthopedics medical device Profemur Stem is the subject of an FDA Class I recall. The recall is the result of the medical device having a high rate of postoperative fractures resulting in the need for revision surgery. Patients implanted with the affected device reportedly experience symptoms including:
- Pain
- Fractures
- Skin Rash
- Metallosis (metal poisoning)
Contact your physician if you are experiencing signs and symptoms of complications. Patients are encouraged to call 1-866-872-0211 with any questions.
Zimmer - M/L Taper Hip Prosthesis with Kinectiv Technology
Problem: Zimmer issued a voluntary recall of 64 lots of the M/L Taper with Kinectiv femoral stems and modular necks after they discovered higher than allowed cytotoxicity levels in the devices. In total, the Class I recall affects 752 implants. The recall stated a “reasonable probability” of adverse biological responses and the need for revision surgery.
Patients implanted with one of the affected devices should contact their healthcare provider if they experience any signs of complications. Patients are also encouraged to report adverse events or side effects to the FDA’s MedWatch Safety Information and Adverse Event Reporting Program.
Zimmer - Versys CoCr Head
The Zimmer Versys CoCr Head device was recalled after Zimmer determined that the packaging in Building 1 was not properly validated. Tests were performed to determine if the packaging was sterile, and while it met acceptable standards, Zimmer decided to recall all lots affected by the improperly validated packaging.
Medical devices that are not sterile or properly packaged can lead to complications and an increased risk of infection.
More Information:
Atrium Medical C-QUR Hernia Mesh
Atrium Medical issued a recall for over 145,000 C-QUR hernia mesh products due to packaging problems. Improper packaging led to some devices sticking to the packaging, which removed the protective coating. The Class II recall included C-QUR products:
- Standard Mesh
- V Patch
- TacShield
- Edge
The protective coating on C-QUR hernia mesh devices is important for patient safety. Damaged coatings increase the risk of infection, adhesions, allergic reactions and pain.
Stryker - Rejuvenate Modular & ABG II Modular-Neck Stems
Stryker issued a voluntary recall of the Rejuvenate Modular and ABG II Modular-neck hip stems after discovering potential risks with the stems. The risks included possible corrosion or fretting at the junction, which could lead to adverse tissue reactions, swelling, and pain in the surrounding areas.
Stryker urges patients with these devices to contact their healthcare provider if they experience any pain, swelling, or other signs of a complication.
Smith & Nephew - R3 Acetabular System Cup
Smith & Nephew have recalled the metal liner or “cup” component of the R3 Acetabular System hip implant. The recall was issued after a “higher than normal” level of problems were reported by patients. The R3 cup component was linked to higher rates of infection, fractures, and dislocation. The defect resulted in a higher percentage of patients requiring revision surgery.
Bard Ventralex Hernia Patch
C. R. Bard recalled a number of Ventralex hernia mesh devices due to a defective design. The defect was linked to serious adverse events, including debilitating complications and reactions, such as:
- Chronic pain
- Infection
- Hernia recurrence
- Mesh migration
- Bowel perforation
DePuy - ASR 300 Acetabular Cup System
The rate of ASR patients who needed a revision surgery was found to be higher than the data previously reported. Patients with the device reportedly experienced symptoms including:
- Pain
- Damage to surrounding tissue
- Loosening of implant
- Failure of implant
- Need for revision surgery
Contact your physician if you are experiencing signs and symptoms of complications. Call theASR helpline for additional information at 888-627-2677.
Zimmer - Durom Prosthesis Cup
The Zimmer Durom Prosthesis Cup, a component of the Metasul LDH Head System, was recalled after it was discovered that the instructions for use were inadequate. Implanting surgeons were advised to stop using the device until appropriate training and instructions could be provided.
More Information:
Call us at (888) 458-6825 or fill out our contact form to discuss your legal options.
The consultation is free and confidential.